render
Opening Files
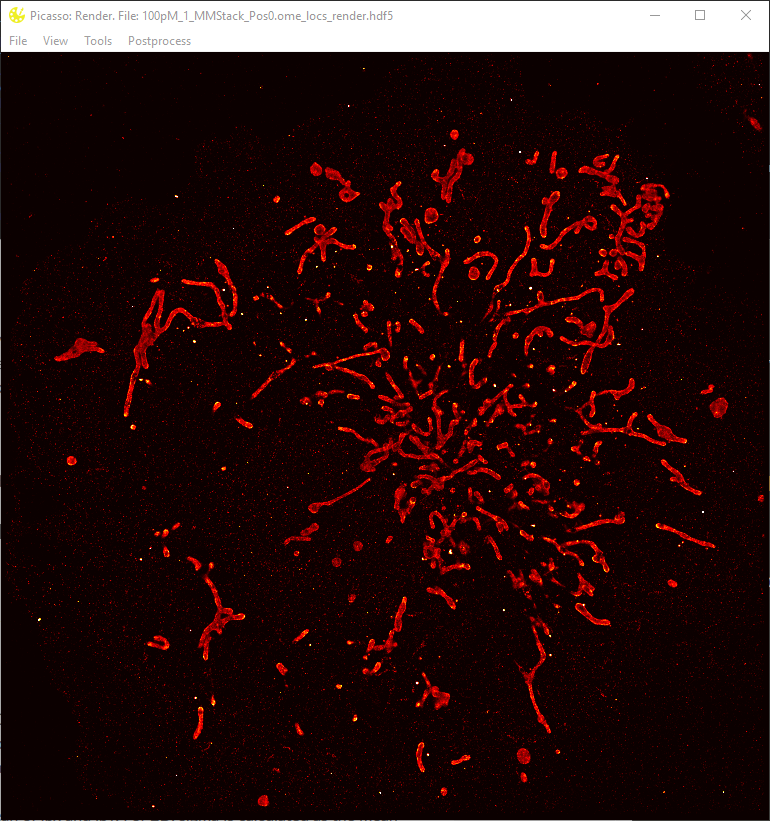
Rendering of the super-resolution image: In
Picasso: Render, open a movie file by dragging a localization file (ending with ‘.hdf5’) into the window or by selectingFile > Open. The super-resolution image will be rendered automatically. A region of choice can be zoomed into by a rectangular selection using the left mouse button. The ‘View’ menu contains more options for zooming and panning.(Optional) Adjust rendering options by selecting
View > Display Settings. The field ‘Oversampling’ defines the number of super-resolution pixels per camera pixel. The contrast settingsMin. DensityandMax. Densitydefine at which number of localizations per super-resolution pixel the minimum and maximum color of the colormap should be applied.(Optional) For multiplexed image acquisition, open HDF5 localization files from other channels subsequently. Alternatively, drag and drop all HDF5 files to be displayed simultaneously.
Drift Correction
Picasso offers three procedures to correct for drift: AIM (Ma, H., et al. Science Advances. 2024., option A), use of specific structures in the image as drift markers (option B) and an RCC algorithm (option C). AIM is precise, robust, quick, requires no user interaction or fiducial markers (although adding them will may improve performance). Although RCC does not require any additional sample preparation, option B depends on the presence of either fiducial markers or inherently clustered structures in the image. On the other hand, option B often supports more precise drift estimation and thus allows for higher image resolution. To achieve the highest possible resolution (ultra-resolution), we recommend AIM or consecutive applications of option C and multiple rounds of option B. The drift markers for option B can be features of the image itself (e.g., protein complexes or DNA origami) or intentionally included markers (e.g., DNA origami or gold nanoparticles). When using DNA origami as drift markers, the correction is typically applied in two rounds: first, with whole DNA origami structures as markers, and, second, using single DNA-PAINT binding sites as markers. In both cases, the precision of drift correction strongly depends on the number of selected drift markers.
Adaptive Intersection Maximization (AIM) drift correction
In
Picasso: Render, selectPostprocess > Undrift by AIM.The dialog asks the user to select:
Segmentation- the number of frames per interval to calculate the drift. The lower the value, the better the temporal resolution of the drift correction, but the higher the computational cost.
Intersection distance (nm)- the maximum distance between two localizations in two consecutive temporal segments to be considered the same molecule. This parameter is robust, 3*NeNA for optimal result is recommended.
Max. drift in segment (nm)- the maximum expected drift between two consecutive temporal segments. If the drift is larger, the algorithm will likely diverge. Setting the parameter up to3 * intersection_distancewill result in fast computation.
After the algorithm finishes, the estimated drift will be displayed in a pop-up window, and the display will show the drift-corrected image.
Marker-based drift correction
In
Picasso: Render, pick drift markers as described in Picking of regions of interest. Use thePick similaroption to automatically detect a large number of drift markers similar to a few manually selected ones.If the structures used as drift markers have an intrinsic size larger than the precision of individual localizations (e.g., DNA origami, large protein complexes), it is critical to select a large number of structures. Otherwise, the statistic for calculating the drift in each frame (the mean displacement of localization to the structure’s center of mass) is not valid.
Select
Postprocess > Undrift from pickedto compute and apply the drift correction.(Optional) Save the drift-corrected localizations by selecting
File > Save localizations.
Redundant cross-correlation drift correction
In
Picasso: Render, selectPostprocess > Undrift by RCC.A dialog will appear asking for the segmentation parameter. Although the default value, 1,000 frames, is a sensible choice for most movies, it might be necessary to adjust the segmentation parameter of the algorithm, depending on the total number of frames in the movie and the number of localizations per frame. A smaller segment size results in better temporal drift resolution but requires a movie with more localizations per frame.
After the algorithm finishes, the estimated drift will be displayed in a pop-up window, and the display will show the drift-corrected image.
Picking of regions of interest
Manual selection. Open
Picasso: Renderand load the localization HDF5 file to be processed.Switch the active tool by selecting
Tools > Pick. The mouse cursor will now change to a circle. Alternatively, openTools > Tools Settingsto change the shape into a rectangle. Lastly, choosingPolygonallows for drawing polygons of any shape.Set the size of the pick circle by adjusting the
Diameterfield in the tool settings dialog (Tools > Tools Settings). Alternatively, chooseWidthfor a rectangular shape.Pick regions of interest using the circular mouse cursor by clicking the left mouse button. All localizations within the circle will be selected for further processing.
(Optional) Automated region of interest selection. Select
Tools > Pick similarto automatically detect and pick structures that have similar numbers of localizations and RMS deviation (RMSD) from their center of mass than already-picked structures. The upper and lower thresholds for these similarity measures are the respective standard deviations of already-picked regions, scaled by a tunable factor. This factor can be adjusted using the fieldTools > Tools Settings > Pick similar ± range. To display the mean and standard deviation of localization number and RMSD for currently picked regions, selectView > Show infoand clickCalculate info below.(Optional) Exporting of pick information. All localizations in picked regions can be saved by selecting
File > Save picked localizations. The resulting HDF5 file will contain a new integer columngroupindicating to which pick each localization is assigned.(Optional) Statistics about each pick region can be saved by selecting
File > Save pick properties. The resulting HDF5 file is not a localization file. Instead, it holds a data set calledgroupsin which the rows show statistical values for each pick region.(Optional) The picked positions and diameter itself can be saved by selecting
File > Save pick regions. Such saved pick information can also be loaded intoPicasso: Renderby selectingFile > Load pick regions.
NOTE: Rectangular picks can be used to generate the projections of localizations onto the rectangle’s axes. To do so, select rectangular picks and save picked localizations (File > Save picked localizations). The resulting hdf5 files will contain columns x_pick_rot and y_pick_rot, which are the projections of localizations onto and against the “drawing” axis of the rectangle, respectively.
3D rotation window
The 3D rotation window allows the user to render 3D localization data. To use it, select a single pick region (Tools > Pick) and click View > Update rotation window. Some of the display settings (colors, blur method, etc.) are automatically uploaded to the rotation window.
The user may perform multiple actions in the rotation window, including: saving rotated localizations, building animations (.mp4 format), rotating by a specified angle, etc.
Note that to build animations, the user must have ffmpeg installed on their system.
Rotation around z-axis is available by pressing Ctrl/Command. Rotation axis can be frozen by pressing x/y/z to freeze around the corresponding axes (to freeze around the z-axis, Ctrl/Command must be pressed as well).
RESI
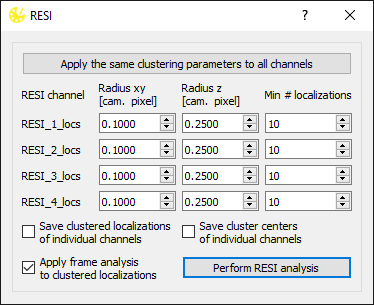
In Picasso 0.6.0, a new RESI (Resolution Enhancement by Sequential Imaging) dialog was introduced. It allows for a substantial resolution boost by sequential imaging of a single target with multiple labels with Exchange-PAINT (Reinhardt, Masullo, Baudrexel, Steen, et al., Nature, 2023. DOI: 10.1038/s41586-023-05925-9).
To use RESI, prepare your individual RESI channels (localization, undrifting, filtering and alignment). Load such localization lists into Picasso Render and open Postprocess > RESI. The dialog shown above will appear. Each channel will be clustered using the SMLM clusterer (other clustering algorithms could be applied as well although only the SMLM clusterer is implemented for RESI in Picasso). Clustering parameters can be defined for each RESI channel individually, although it is possible to apply the same parameters to all channels by clicking Apply the same clustering parameters to all channels, which will copy the clustering parameters from the first row and paste it to all other channels.
Next, the user needs to specify whether or not to save clustered localizations or cluster centers from each of the RESI channels individually, and whether to apply basic frame analysis (to minimize the effect of sticking events). For the explanation of the parameters, see SMLM clusterer below.
Upon clicking Perform RESI analysis, each of the loaded channels is clustered, cluster centers are extracted and combined from all RESI channels to create the final RESI file.
G5M
In Picasso 0.9.5, a new algorithm for molecular mapping (i.e., finding the positions of individual molecules from localizations) was introduced: G5M (Gaussian Mixture Modeling with Modifications for Molecular Mapping; Kowalewski, Reinhardt et al. Nature Comms, 2026. DOI: 10.1038/s41467-026-70198-5). G5M is based on Gaussian Mixture Modeling (GMM) but includes several modifications to make it suitable for molecular mapping. All the technicalities as well as the user guide of the method are explained in the publication mentioned and its Supplementary Information. Please refer to picasso.g5m for the details of the implementation. Below is a brief summary of the user guide.
G5M requires some preprocessing of localizations to filter out the badly fitted ones, especially the ones arising from crosstalk (overlapping blinking). These can be excluded from 2D data where the ellipticity and size of the image of an emitter in x and y can be filtered (in Picasso these are found under names “ellipticity”, “sx” and “sy”, respectively). Moreover, the photon count can be cut-off as crosstalk is likely to result in a higher-intensity signal. In 3D data these filters are less reliable due to astigmatism, however, “d_zcalib” could be used. We strongly encourage avoiding dense blinking, where emission signals from neighboring molecules overlap, especially during 3D image acquisition.
Prior to molecular mapping, clustering of localizations is required to split the data into smaller chunks. For many datasets, DBSCAN works well. While in some cases some adjustments may be needed, we recommend the following DBSCAN parameters: In 2D, DBSCAN radius (epsilon) of 2*LP, in 3D - 3*LP (LP - average localization precision of the dataset, for example, NeNA or median localization precision). Default min. samples is set to 4. Clustering in Picasso adds the group column to the localization file, which is required for G5M. Note: G5M relies on the information in the ``group`` column, therefore, if it is overwritten (for example, by picking localizations after DBSCAN clustering), G5M will not work.
To account for fluorophore non-specific sticking, frame analysis is normally recommended (especially the filtering of st. dev. of frame per molecule). However, if localizations from neighboring localization clouds overlap, this is not sufficient due to ambigous assignment of localizations to molecules. Therefore, we recommend filtering of molecules that express too few binding events (saved in the column n_events). In the publication, we recommend a threshold of at least 3 binding events per molecule.
The final postprocessing step is log-likelihood filtering (using the column p_val). The recommended threshold is > 0.0015, however, it might need to be adjusted for your data, especially in 3D this can be too conservative.
As a final check for overfitting (i.e., too many assigned molecules), G5M automatically saves a bar plot of the number of binding events per molecule (n_events column) for clustered (with neighbors within 25 nm) and sparse (without neighbors within 80 nm) molecules. If the clustered molecules show fewer binding events that the sparse molecules, overfitting likely occurred. See Fig. S15 of the publication for an example of well-behaved data. As of v0.9.8, Picasso saves the plot showing relative σ values (i.e., the fitted Gaussian σ divided by the average loc. precision around the molecule). This can be used to estimate if the loc. precision values are accurate (if not, many molecules will have relative σ values close to the user-selected min./max. σ). As of version 0.9.10, Picasso runs KS 2 sample test to compare the two distributions. The output test statistic and theoretical p value correspond to the KS test, while the permutation p value is calculated by randomly permuting the labels of clustered and sparse molecules 1,000 times and calculating the fraction of permutations that result in a KS test statistic as extreme as the one observed with the original labels.
If the outcome of G5M seems unsatisfactory, please check the following:
Make sure that
groupcolumn is present in the localization file and contains the correct information (i.e., from DBSCAN clustering, not from picking localizations);Make sure that the loc. precision values (columns
lpx,lpy,lpz) are correct, comparing NeNA and median loc. precision is a reasonable proxy (without fiducial markers); the most common issue is a miscalibrated camera, leading to incorrect photon counts and thus incorrect loc. precisions;Another reason why the loc. precision values can be off is due to the small box size in the localization step; especially in 3D astigmatic imaging, single-emitter images can be quite large, potentially exceeding the user-defined box size; in such cases, we recommend increasing the box size in the localization step and rerunning the analysis;
Inspect if the localizations were preprocessed as described above;
Rerun the analysis without postprocessing (filtering) and redo it manually, since some steps may be too stringent, such as
p_valorn_events(latter especially for short acquisition times);Adjust min./max. σ, especially too low max. σ may lead to high false positive error rates (i.e., overfitting); We suggest inspecting
rel_sigmavalues of the assigned molecules, which are calculated as the fitted σ divided by the mean localization precision of the surrounding localizations. If the values are close to the user-selected min./max. σ, min./max. σ might need to be adjusted. Alternatively, this might be a sign of inaccurate/inprecise loc. precision values, see above;Adjust min. locs;
Adjust DBSCAN (or other clustering algorithm) parameters. For example, if G5M takes too long to run, the DBSCAN clusters most likely contain too many molecules. In such a case, we recommend splitting such clusters further;
Dialogs
Display Settings
Allows to change the display settings. Open via View > Display Settings.
General
Adjust the general display settings.
Zoom
Set the magnification factor.
Oversampling
Set the oversampling. Choose dynamic to automatically adjust to current window size when zooming.
Minimap
Click show minimap to display a minimap in the upper left corner to localize where the current field of view is within the image.
Contrast
Define the minimum and maximum density of the and select a colormap. Over 100 colormaps are available. The last option Custom requires the user to load their own .npy file containg a numpy array with a custom colormap. The selected colormap will be saved when closing render.
Blur
Select a blur method. Available options are: * None * One-Pixel-Blur * Individual Localization Precision * Individual Localization Precision, iso
Camera
Select the pixel size of the camera. This will be automatically set to a default value or the value specified in the *.yaml file.
Scale Bar
Activate scale bar. The length of the scale bar is calculated with the Pixel Size set in the Camera dialog. Activate Print scale bar length to additionally print the length.
Render properties
This allows rendering properties by color.
Show Info
Displays the info dialog.
Display
Shows the image width/height, the coordinates, and dimensions of the current FoV.
Movie
Displays the median fit precision of the dataset. Clicking on Calculate allows calculating the precision via the NeNA approach. See DOI: 10.1007/s00418-014-1192-3.
FRC
Displays the FRC resolution of the dataset. Takes in the image in the current FOV and calculates the FRC resolution via splitting the localizations into two halves. Based on the approach from 10.1038/nmeth.2448. Does not take into account the Q factor for multiple blinking.
Field of view
Shows the number of localizations in the current FoV.
Picks
Allows calculating statistics about the picked localizations. Press Calculate info below to calculate. Ignore dark times allows treating consecutive localizations as on, even if there are localizations (specified by the parameter) missing between them. When defining the number of units per pick, you can calibrate the influx rate via Calibrate influx. A histogram of the dark and bright time can be plotted when clicking Histograms.